The data in this experiment are a sample of 400 controls from the European Prospective Investigation of Cancer (EPIC) Norfolk study (http://www.srl.cam.ac.uk/epic/) each with approximately 250k Perlegen (http://www.perlegen.com/) SNPs (the EPIC 400 study). This is part of a screen sample from a multistage study of breast cancer in EPIC.
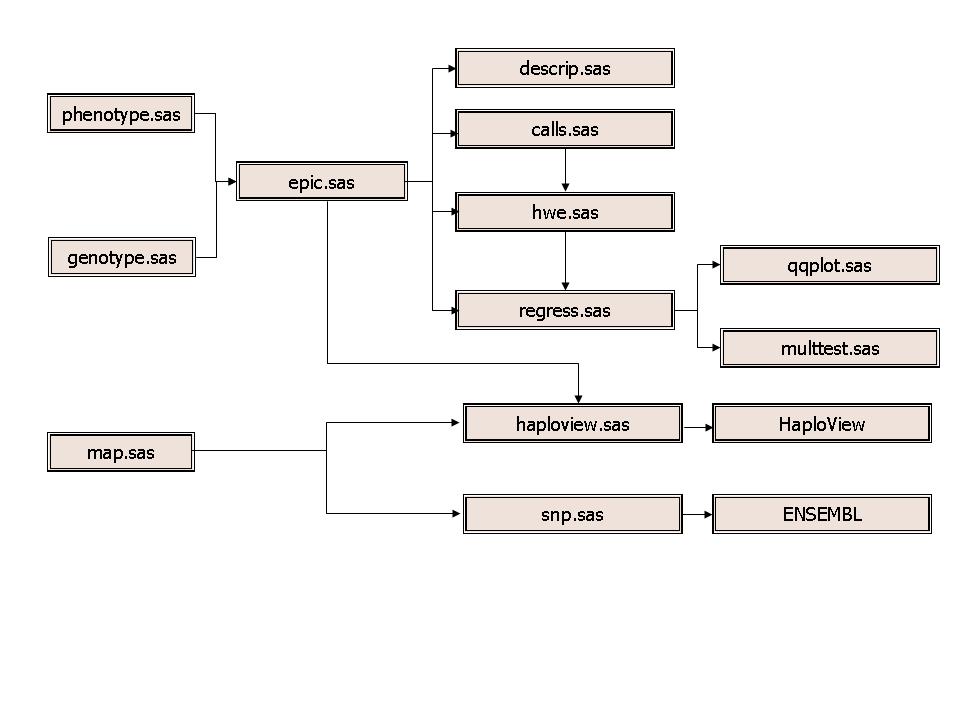
The flowchart of our implementation is shown in Figure 1. The input data have three sources of information, i.e., genotype, map and phenotype. The genotype data contain actual genotypes for all individuals in the so-called long format (individual ID, SNP ID, and genotype). Map information show position of each SNP by chromosome. Phenotypic information is in the usual tabular format (individual ID, sex, body mass index and other measurements). These three sources of information are merged into a combined dataset for analysis. For screen purpose, only single point analysis was conducted. Call rates were obtained, as with HWE tests for all SNPs, the results of which were used as an inclusion/exclusion filter of SNPs in the regression analysis. The code for SNP information including HWE tests is shown as follows,
proc sort
data=epic;
by chr
pos;
run;
ods select
none;
proc allele
data=epic genocol;
ods output
markersumm=ms allelefreq=af genotypefreq=gf;
by chr
pos;
var
a1a2;
run;
ods select all;
The input data is sorted by chromosome (chr) and positions (pos), as input to PROC ALLELE which accepts genotype (genocol) and outputs summary information of SNPs (markersumm), allele frequencies (allefreq) and genotype frequencies (genotypefreq). The outputs are stored in ODS (output delivery system) databases by chromosomes and SNP positions, and all outputs for individual SNPs are suppressed (ods select none). This shows great simplicity.The raw genotype data and map information can be used to construct input files for HAPLOVIEW (http://www.broad.mit.edu/mpg/haploview/) for visualisation. The SNPs involved can be submitted to ENSEMBL (http://www.ensembl.org/index.html) to obtain gene annotation.
Several useful features are notable in this analysis.
First, we do not require any other software to manage data. In a traditional
statistical analysis, the data usually takes the so-called wide format, where
rows indicate sample and columns variables. Since the number of SNPs is quite
large, it is more sensible to organise genotype data into the so-called long
format with only a few columns indicating individual IDs, SNP IDs and genotypes.
Although this requires larger amount of storage but the analysis is considerable
simpler, for one can perform analysis for each SNP in sequence and store the
results in a systematic fashion. Second, we were able to take advantage of
SAS/GENETICS module for HWE and haplotype analysis. Third, all the outputs are
available as databases for re-use and it is possible to generate data for
external software programs such as HAPLOVIEW. The facility of result database is
possible with ODS. Lastly, the SAS programs we developed can run without
c
We have kept the
comma-separated data in compressed format, to be readily processed by pipe
mec
Figure 1. A flowchart of the EPIC 400 Analysis. The raw data consist of genotypes, phenotypes and map information, to be merged. Descriptive statistics are then obtained, followed by calculation of call rates and HWE, the results of which are fed into the regression which assesses statistical significance and comparison with theoretical distribution by Q-Q plot. The raw data can also be reformatted into HAPLOVIEW input files so that specific region in the genome can be visualised.